新靶點!廣州醫(yī)科大學代小艷團隊揭示Par3L在動脈粥樣硬化中的作用及機制
動脈粥樣硬化性心血管疾病(ASCVD)已成為全球發(fā)病率和死亡率的主要原因�����,研究表明M1巨噬細胞極化參與動脈粥樣硬化的病理生理�����,抑制M1巨噬細胞極化可減輕動脈粥樣硬化��。Par3L蛋白是Par3家族的一種新型同源蛋白�����,參與細胞極性的建立�,已被證明可以維持乳腺干細胞的干性,促進結直腸癌細胞的存活�。然而,極性蛋白Par3L是否以及如何影響動脈粥樣硬化仍然未知���。
2023年9月���,廣州醫(yī)科大學代小艷教授團隊在Acta Pharmacologica Sinica(IF8.2)發(fā)表題為“Par3L, a polarity protein, promotes M1 macrophage polarization and aggravates atherosclerosis in mice via p65 and ERK activation”的文章�����。研究闡明了Par3L促進M1巨噬細胞極化并加重動脈粥樣硬化的作用�����,揭示Par3L可能是動脈粥樣硬化的潛在治療靶點���。
研究方法與結果
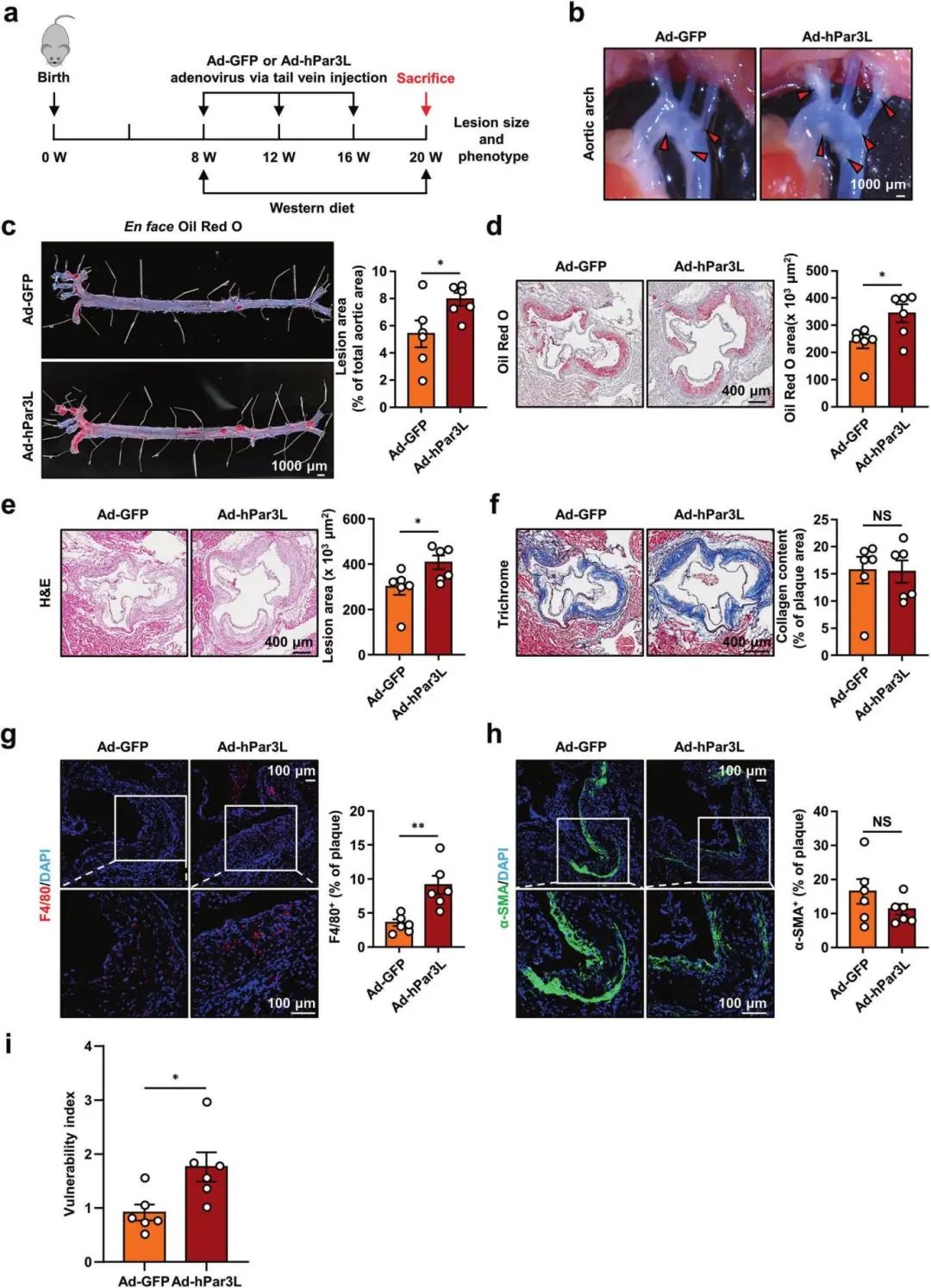
1��、腺病毒介導的Par3L過表達加重Apoe?/?小鼠體內(nèi)動脈粥樣硬化
數(shù)據(jù)表明Par3L在人和小鼠動脈粥樣硬化斑塊中表達顯著上調�,并在原代小鼠巨噬細胞中受氧化型低密度脂蛋白oxLDL誘導上調�。為進一步評估Par3L對小鼠動脈粥樣硬化發(fā)展的影響���,作者將Ad-GFP或Ad-hPar3L通過尾靜脈注入Apoe?/?小鼠體內(nèi)�����,隨后給予12周西方飲食喂養(yǎng)����。與Ad-GFP Apoe?/?小鼠相比,Par3L過表達后主動脈粥樣硬化病變面積和斑塊不穩(wěn)定性明顯增加���,同時巨噬細胞的積累顯著增加。這些數(shù)據(jù)表明Par3L加速Apoe-/-小鼠體內(nèi)動脈粥樣硬化斑塊的形成�����。
腺病毒介導的Par3L過表達加重Apoe?/?小鼠動脈粥樣硬化
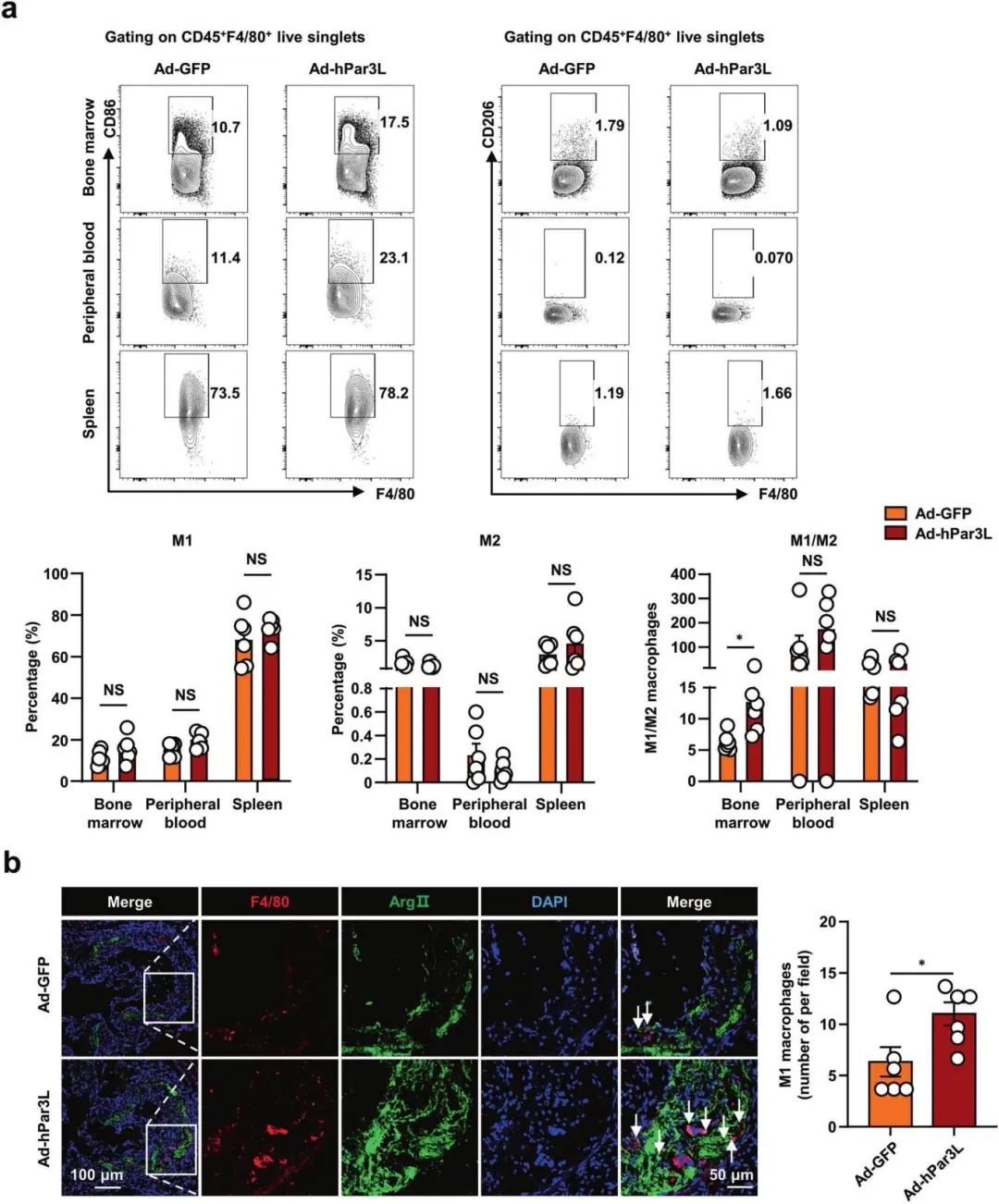
2���、腺病毒介導的Par3L過表達調節(jié)Apoe?/?小鼠體內(nèi)M1巨噬細胞極化
巨噬細胞已被證明參與動脈粥樣硬化的發(fā)病過程�。經(jīng)典活化的M1巨噬細胞參與引發(fā)和維持炎癥�����,而交替活化的M2巨噬細胞與消除炎癥有關��。流式細胞術評估發(fā)現(xiàn)Par3L明顯增加了M1 / M2巨噬細胞的比例��,表明Par3L參與了M1巨噬細胞的極化���,免疫熒光分析表明相對于Ad-GFP組���,Ad-hPar3L組小鼠動脈粥樣硬化病變中M1巨噬細胞的積累更多。這些數(shù)據(jù)表明Par3L促進Apoe?/?小鼠體內(nèi)M1巨噬細胞極化��。進一步檢測發(fā)現(xiàn)Par3L促進LPS和IFNγ誘導的M1巨噬細胞體外極化��。
腺病毒介導的Par3L過表達調節(jié)Apoe?/?小鼠體內(nèi)M1巨噬細胞極化
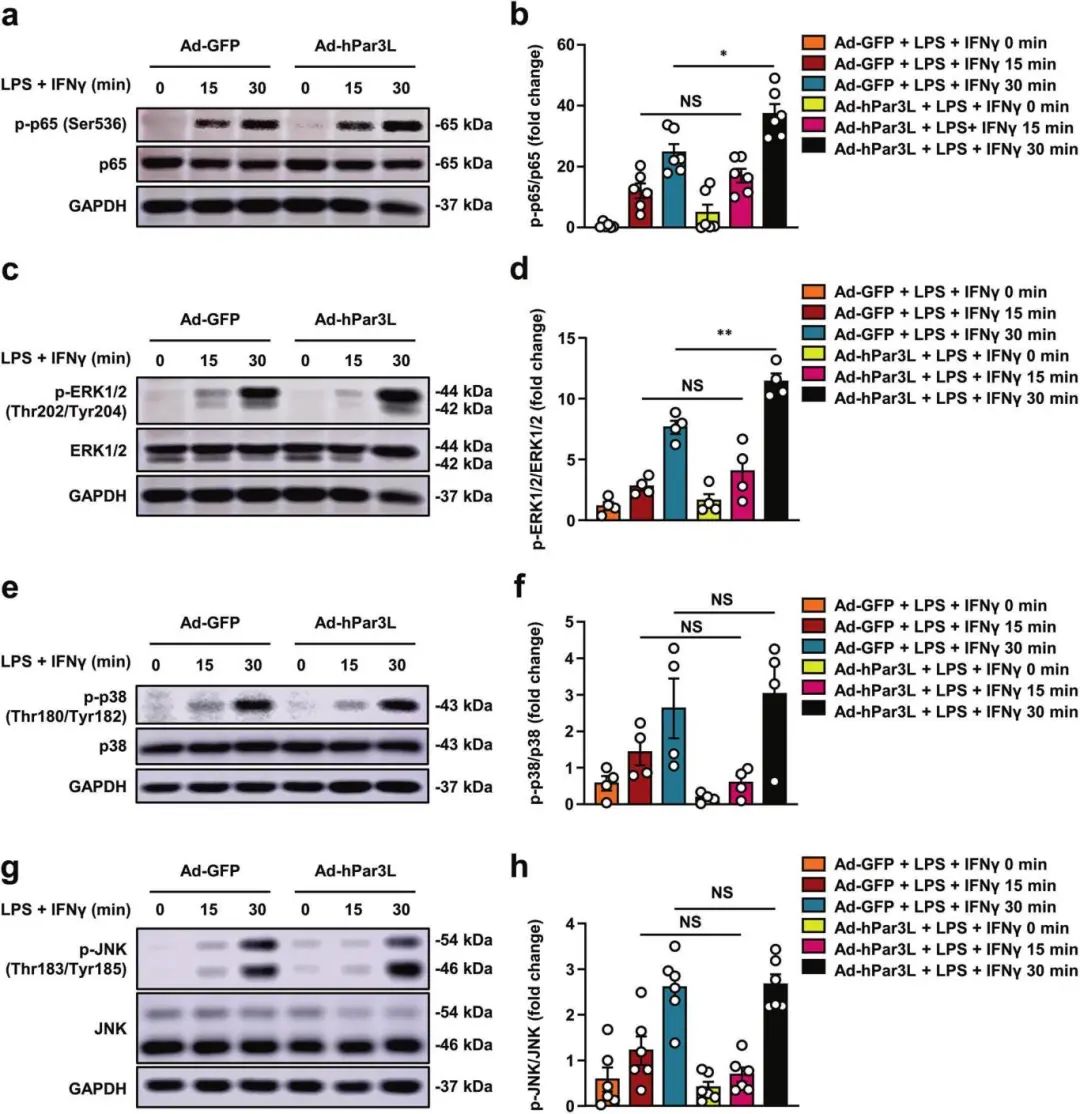
3��、Par3L過表達通過激活p65和ERK信號誘導M1巨噬細胞極化
研究報道NF-κB p65亞基的激活通過TLR配體調節(jié)M1巨噬細胞極化�����,產(chǎn)生促炎因子�,1,2 -乙二胺SQ109通過p38 MAPK途徑促進M1巨噬細胞極化����。鑒于NF-κB和MAPK在M1巨噬細胞極化中的關鍵作用����,作者試圖確定Par3L是否影響這些途徑�����。結果顯示�,在LPS和IFNγ處理的腹腔巨噬細胞(PMs)中,Par3L過表達增加了p-p65和p-ERK1/2���,而在感染Ad-GFP或Ad-hPar3L的PMs中�,p-p38和p-JNK沒有明顯變化�。綜上所述,這些數(shù)據(jù)表明Par3L過表達通過激活p65和ERK信號誘導M1巨噬細胞極化����。進一步驗證表明阻斷NF-κB和ERK信號通路可消除Par3L對M1巨噬細胞極化的影響�����。
Par3L過表達通過激活p65和ERK信號誘導M1巨噬細胞極化
結論
本研究證明Par3L通過激活NF-κB和ERK通路誘導巨噬細胞M1極化并加重動脈粥樣硬化���,揭示Par3L是M1巨噬細胞極化的重要內(nèi)源性調節(jié)因子�����,也是ASCVD的一個有希望的治療靶點��。


 當前位置:首頁 > 新聞中心 > 新聞資訊
當前位置:首頁 > 新聞中心 > 新聞資訊