Advanced Science|山東大學(xué)馬春紅教授團(tuán)隊(duì)揭示Tipe1是胰島β細(xì)胞穩(wěn)態(tài)及T2D發(fā)病的關(guān)鍵調(diào)控因子
2024年2月28日�,山東大學(xué)基礎(chǔ)醫(yī)學(xué)院馬春紅��、高立芬研究團(tuán)隊(duì)在Advanced Science(中科院一區(qū)TOP/JCR Q1��,5年IF=16.7)上發(fā)表題為“Beta-Cell Tipe1 Orchestrates Insulin Secretion and Cell Proliferation by Promoting Gαs/cAMP Signaling via USP5”的研究論文����,揭示了Tipe1分子調(diào)控胰島β細(xì)胞穩(wěn)態(tài)抑制糖尿病進(jìn)展的新型生物學(xué)功能�,Tipe1通過去泛素酶USP5抑制k48連接的Gαs的泛素化降解進(jìn)而調(diào)控β細(xì)胞的增殖和功能,表明Tipe1可能是T2D干預(yù)的新治療靶點(diǎn)�。


01研究背景
胰腺β細(xì)胞在維持胰島素分泌和葡萄糖穩(wěn)態(tài)中起著至關(guān)重要的作用�,β細(xì)胞質(zhì)量和胰島素分泌不足是2型糖尿病(T2D)發(fā)展的必要條件���。TNF-α誘導(dǎo)的蛋白8-like 1 (Tipe1)在多種疾病中起著至關(guān)重要的作用�,并在正常小鼠的胰島中高度表達(dá)�����,然而其在T2D發(fā)病機(jī)制中的特定作用仍未被廣泛探索���。
02研究結(jié)果
1����、β細(xì)胞中Tipe1敲低導(dǎo)致小鼠嚴(yán)重的糖尿病表型
作者研究發(fā)現(xiàn)在生理?xiàng)l件下��,胰島β細(xì)胞中Tipe1的表達(dá)與關(guān)鍵胰島素相關(guān)基因的表達(dá)呈正相關(guān)�。為評估Tipe1在β細(xì)胞中的作用���,作者構(gòu)建了β細(xì)胞特異性Tipe1缺陷(Ins2-Tipe1BKO)小鼠���。與對照組相比,Ins2-Tipe1BKO小鼠胰島中Tipe1的表達(dá)明顯降低,實(shí)驗(yàn)表明β細(xì)胞中Tipe1的缺乏會損害小鼠葡萄糖穩(wěn)態(tài)����。作者進(jìn)一步構(gòu)建了β細(xì)胞條件性敲除Tipe1的db/db小鼠,即Ins2-Tipe1BKO-db/db小鼠�����,研究發(fā)現(xiàn)Tipe1條件性缺乏會導(dǎo)致db/db小鼠胰島素分泌不足。有證據(jù)表明���,糖尿病前期或糖尿病患者的特點(diǎn)是呼吸交換率(RER)降低和代謝不靈活,為了研究Ins2Tipe1BKO-db/db小鼠比db/db小鼠血糖水平更高��、體重更輕的原因����,通過檢測兩類小鼠食物攝入量����、飲料攝入量和能量消耗以及胰島變化,發(fā)現(xiàn)db/db小鼠β細(xì)胞中Tipe1的缺乏加速了T2D條件下β細(xì)胞損失和胰島素不足����。此外���,作者發(fā)現(xiàn)Tipe1的缺失降低了β細(xì)胞在HFD刺激下的代償性增殖,導(dǎo)致β細(xì)胞質(zhì)量不足����。
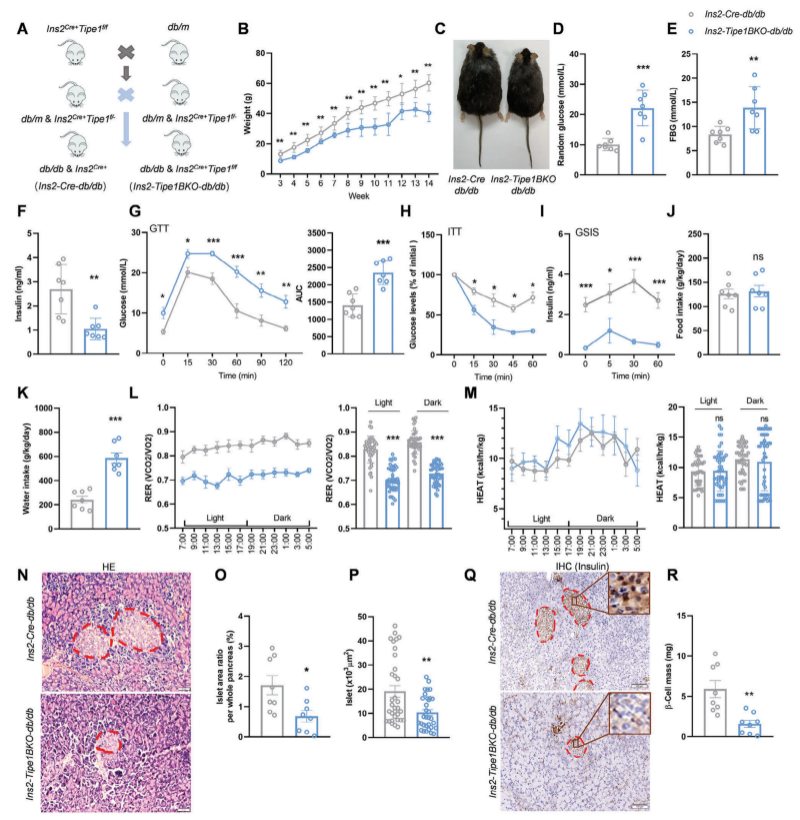
圖1. 在db/db小鼠中�����,β細(xì)胞中Tipe1敲低導(dǎo)致嚴(yán)重的糖尿病表型
2����、Tipe1促進(jìn)β細(xì)胞增殖和胰島素分泌
為了更好地了解β細(xì)胞中Tipe1缺失的影響,作者使用分離的胰島和MIN6 β細(xì)胞系來檢查Tipe1是否可以以細(xì)胞自主的方式調(diào)節(jié)β細(xì)胞功能��。從Ins2-Tipe1BKO和Ins2-Cre小鼠中分離出原代胰島�,在體外條件下用不同濃度葡萄糖處理�����,發(fā)現(xiàn)Ins2-Tipe1BKO小鼠胰島細(xì)胞內(nèi)胰島素水平降低�����。此外�����,當(dāng)用較高濃度葡萄糖刺激胰島時(shí)�����,Ins2-Tipe1BKO小鼠的胰島素分泌明顯減少����。與Ins2-Cre小鼠相比�����,Ins2-Tipe1BKO小鼠胰島中胰島素相關(guān)基因的表達(dá)一致降低��。在MIN6細(xì)胞中敲低或增加Tipe1的表達(dá)��,發(fā)現(xiàn)Tipe1敲低的MIN6細(xì)胞增殖下降�,而Tipe1過表達(dá)的MIN6細(xì)胞增殖增加����。通過分離Ins2-Tipe1BKO和對照小鼠原代胰島�,發(fā)現(xiàn)Tipe1敲除小鼠胰島中的增殖相關(guān)標(biāo)志物減少���。有證據(jù)表明�����,長時(shí)間(3-6個(gè)月)暴露于高脂環(huán)境可以增加β細(xì)胞的增殖��。與對照組相比�����,Ins2-Tipe1BKO-HFD和Ins2-Tipe1BKO-db/db小鼠的β細(xì)胞增殖顯著降低���。作者進(jìn)一步利用腺病毒進(jìn)行Tipe1拯救實(shí)驗(yàn)發(fā)現(xiàn)Tipe1過表達(dá)可以逆轉(zhuǎn)其缺失引起的表型,表明Tipe1以細(xì)胞自主的方式促進(jìn)β細(xì)胞增殖和胰島素分泌���。
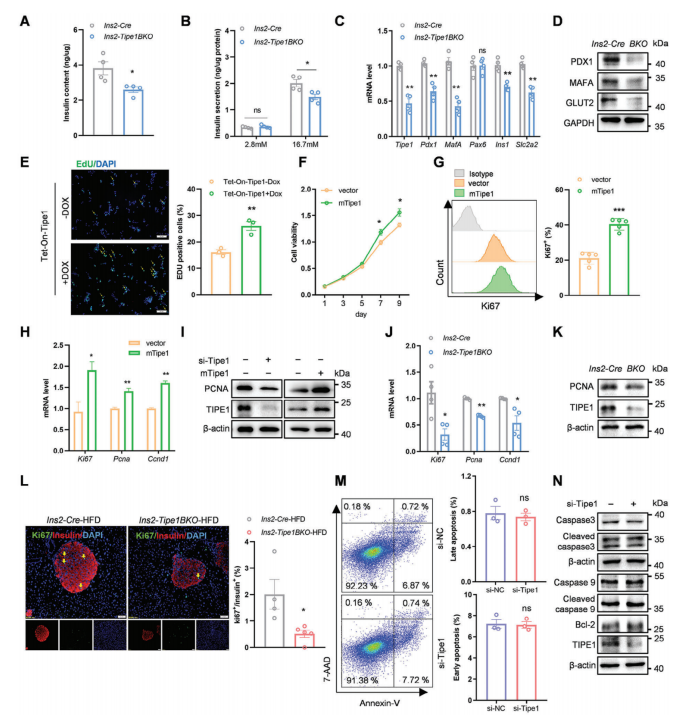
圖2. Tipe1以細(xì)胞自主的方式促進(jìn)β細(xì)胞增殖和胰島素分泌
3�����、Tipe1通過Gαs/cAMP通路維持β細(xì)胞功能
接下來�����,作者進(jìn)一步探究了Tipe1調(diào)節(jié)胰島β細(xì)胞的分子機(jī)制�,利用IP/質(zhì)譜法��,在β細(xì)胞中發(fā)現(xiàn)了大量潛在的Tipe1相互作用蛋白���,并對已報(bào)道的人類IGT(糖耐量受損)和T2D胰島(GSE50398)中不同表達(dá)的基因進(jìn)行聚類分析��。根據(jù)分析結(jié)果���,作者進(jìn)一步探究了Tipe1與Gαs(由Gnas編碼)或GΑSP1(由Gprasp1編碼)的相互作用。同時(shí)����,共聚焦實(shí)驗(yàn)進(jìn)一步驗(yàn)證了Tipe1和Gαs在MIN6細(xì)胞中的共定位����。為了證實(shí)Gαs是否與Tipe1介導(dǎo)的胰島β細(xì)胞調(diào)控有關(guān),將過表達(dá)Gnas的腺病毒轉(zhuǎn)導(dǎo)Tipe1沉默的MIN6細(xì)胞和Ins2-Tipe1BKO小鼠的胰島�����,發(fā)現(xiàn)Gnas的過表達(dá)逆轉(zhuǎn)了MIN6細(xì)胞的增殖。此外�����,在Ins2-Tipe1BKO和Ins2-Tipe1BKO-db/db小鼠的胰島中,高糖刺激的胰島素分泌也被Gnas過表達(dá)所逆轉(zhuǎn)�,表明Gαs參與Tipe1介導(dǎo)的β細(xì)胞調(diào)控。作者進(jìn)一步研究發(fā)現(xiàn)Tipe1通過Gαs/cAMP途徑調(diào)節(jié)β細(xì)胞增殖和胰島素分泌��。
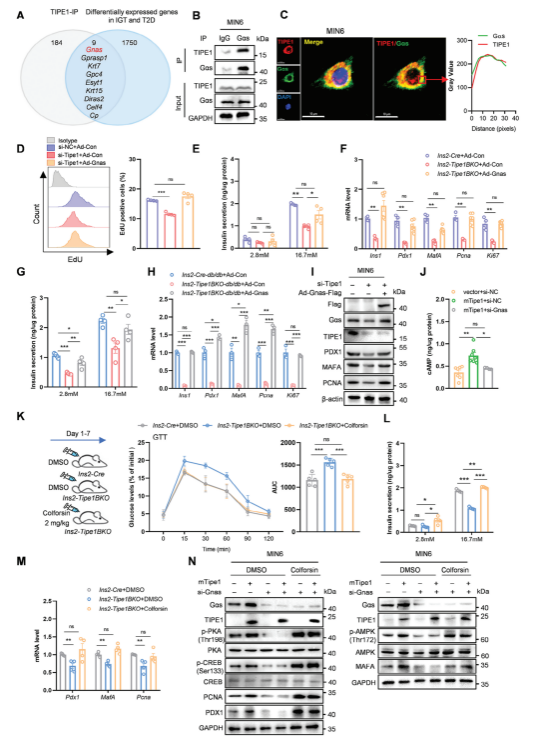
圖3. Tipe1通過Gαs/cAMP通路調(diào)控β細(xì)胞功能
4、Tipe1通過抑制k48連接的泛素化降解來穩(wěn)定Gαs
作者進(jìn)一步證實(shí)了Tipe1與Gαs的相互作用�����,并且發(fā)現(xiàn)Tipe1通過阻斷Gαs蛋白酶體降解來增強(qiáng)Gαs的豐度���。通過對與Tipe1存在潛在相互作用的蛋白進(jìn)行分析�,發(fā)現(xiàn)這些蛋白參與了RNA剪接��、蛋白質(zhì)轉(zhuǎn)運(yùn)和蛋白質(zhì)泛素化過程的調(diào)節(jié)���,有報(bào)道稱,Tipe1可以抑制其結(jié)合蛋白的多泛素化����,Gαs在K28位點(diǎn)有乙酰化修飾��,但沒有證據(jù)表明Gαs的泛素化修飾����。然后���,作者試圖確定Tipe1是否可以調(diào)節(jié)Gαs的泛素化修飾��。首先檢測了內(nèi)源性Gαs在HEK293T和MIN6細(xì)胞中的泛素化積累���,發(fā)現(xiàn)免疫沉淀的Gαs蛋白存在泛素化修飾���,同時(shí)Tipe1過表達(dá)抑制內(nèi)源性和外源性Gαs蛋白的泛素化積累���,而Tipe1敲低可促進(jìn)Gαs泛素化。一致地�,Tipe1過表達(dá)抑制了MIN6細(xì)胞中k48連接的Gαs泛素化���,而敲低Tipe1逆轉(zhuǎn)了上述作用。進(jìn)一步確定了Gαs的泛素化位點(diǎn),過表達(dá)Tipe1降低了Gαs在8�����、28和96位的泛素化�����,這表明Gαs的三個(gè)賴氨酸殘基(K8��、K28和K96)負(fù)責(zé)Tipe1介導(dǎo)的Gαs多泛素化��。進(jìn)一步研究表明Tipe1通過USP5抑制Gαs泛素化降解���。

圖4. Tipe1通過抑制k48連接的泛素化來穩(wěn)定Gαs
03小結(jié)
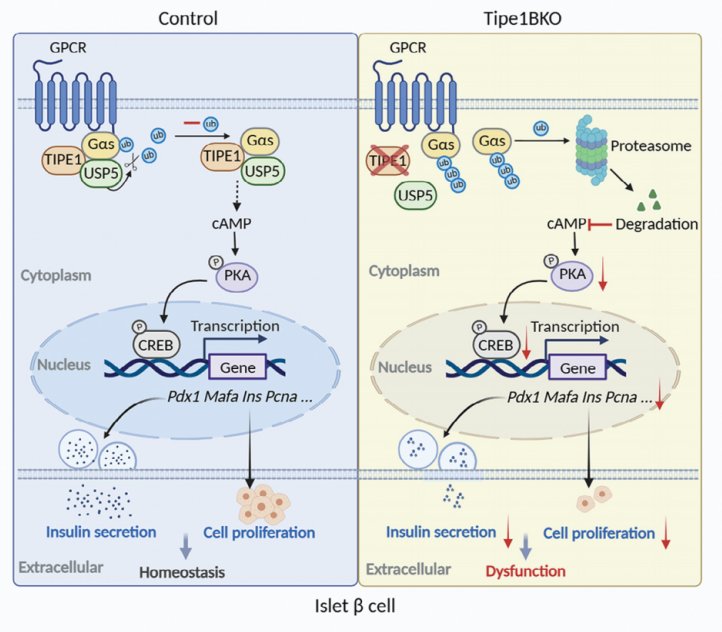
本研究確定了Tipe1在調(diào)節(jié)β細(xì)胞穩(wěn)態(tài)中的關(guān)鍵作用���,首次報(bào)道了Tipe1調(diào)節(jié)Gαs的泛素化修飾,并通過去泛素化酶USP5抑制Gαs的多泛素化��,從而穩(wěn)定Gαs�,提高了cAMP的下游水平����,表明Tipe1可能成為T2D干預(yù)的新靶點(diǎn)。
*山東大學(xué)基礎(chǔ)醫(yī)學(xué)院博士研究生丁璐為該論文的第一作者�,基礎(chǔ)醫(yī)學(xué)院高立芬教授為該論文通訊作者,山東大學(xué)為該論文的第一作者單位和通訊作者單位�。













